Quick Buzz Feed

Proteins are highly diverse functional polymers where the specific sequence of amino acids, selected from a standard genetically-encoded alphabet of twenty (C20), determines the structure and ultimately the function of the resulting folded protein. This standard alphabet has been identified to be non-randomly distributed in physicochemical properties crucial to both structure-formation and function, often referred to as coverage theory.
Proteins are fundamental functional polymers that exhibit immense diversity, with their specific three-dimensional structure and ultimate function being determined by the unique sequence of amino acids. These amino acids are typically chosen from a standard, genetically-encoded alphabet of twenty (C20). This conventional amino acid set is recognized for its non-random distribution of physicochemical properties, which are critical for both the formation of protein structures and their biological functions, a concept often referred to as 'coverage theory.' This foundational understanding is crucial for any advanced protein design endeavor.
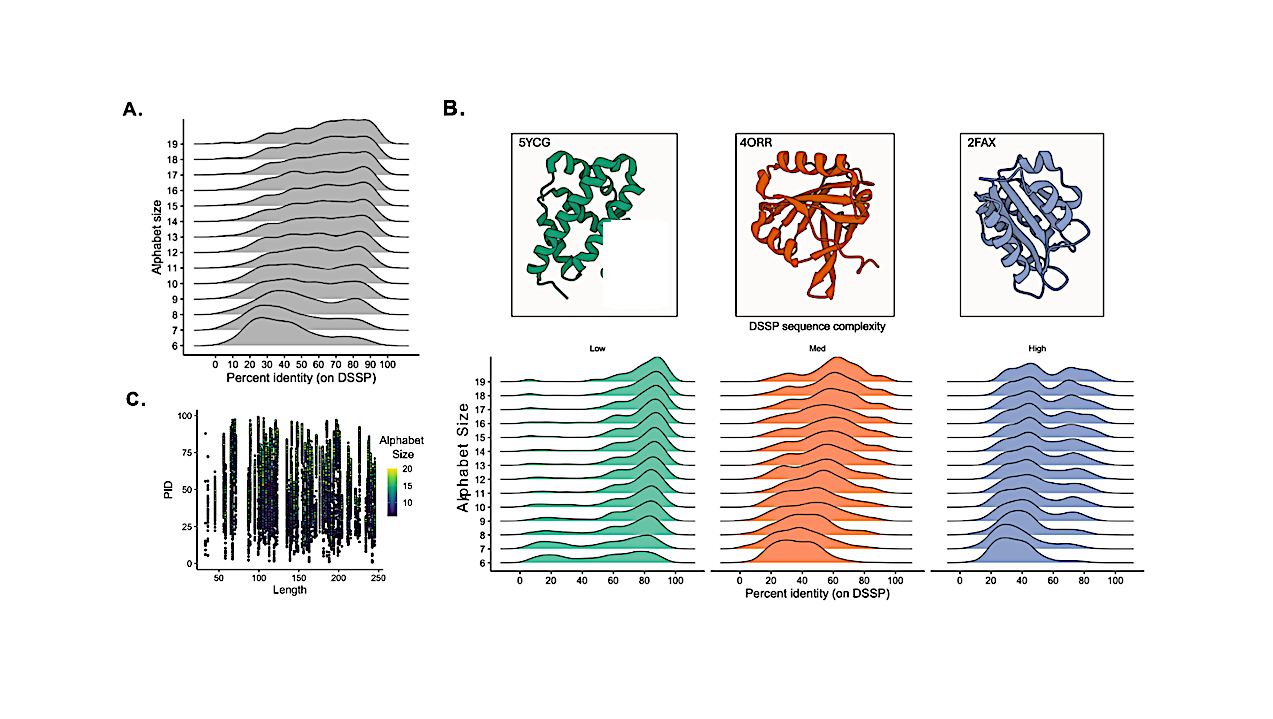
While machine learning models have achieved remarkable advancements in predicting protein structures with high accuracy, the field of de novo protein design, which involves creating novel proteins from scratch, has not yet seen a similar level of development. This research aims to bridge this gap by integrating established contemporary biological theories with cutting-edge advancements in artificial intelligence (AI). A generative AI model has been developed and thoroughly evaluated specifically for protein design. This model was trained using an extensive dataset comprising hundreds of thousands of proteins sourced from the RSCB PDB database, with the goal of designing peptides with custom secondary structure motifs using reduced amino acid alphabets.
The generative AI model demonstrated significant success in its primary objective: designing novel proteins that successfully incorporate desired secondary structure motifs. This capability was observed across a broad spectrum of reduced amino acid alphabets, indicating the model's adaptability and effectiveness. An particularly interesting and unexpected outcome was the tool's frequent ability to capture the full three-dimensional tertiary structure of a target protein. This occurred despite the model being trained exclusively on physicochemical sequence space and DSSP secondary structure data, suggesting that the AI implicitly learned more complex folding rules beyond its explicit training.
The creation of this innovative AI model is expected to significantly advance research across multiple scientific disciplines. Beyond its direct contributions to general scientific AI/machine learning architecture development, it holds substantial implications for protein design within biotechnology, potentially enabling the engineering of proteins with tailored functions for medical, industrial, or environmental applications. Furthermore, this research is relevant to astrobiology, offering insights into how life might evolve or be synthetically constructed in extraterrestrial environments with different biochemical constraints, and to early-Earth evolutionary biology, by exploring the potential roles of simpler amino acid sets in the origin of life.
The technical foundation of this work involves a bLSTMa encoder-decoder model architecture. The encoder block, a critical component, is primarily constructed from LSTM (Long Short-Term Memory) encoder layers and incorporates a multi-head self-attention mechanism, which allows the model to weigh the importance of different parts of the input sequence. The model's head, which processes the output from the decoder, channels this information separately through a classifier to predict sequences and a continuous value sequence to forecast associated physicochemical properties. This sophisticated architecture underpins the AI's ability to learn and generate complex protein designs.